Tralokinumab for severe, uncontrolled asthma (STRATOS 1 and STRATOS 2): two randomised, double-blind, placebo-controlled, phase 3 clinical trials
Artículo seleccionado
Tralokinumab for severe, uncontrolled asthma (STRATOS 1 and STRATOS 2): two randomised, double-blind, placebo-controlled, phase 3 clinical trials.
Panettieri RA, Sjöbring U, Péterffy A, Wessman P, Bowen K, Piper E, Colice G, Brightling CE.
LancetRespirMed. 2018 Jul;6(7):511-525. doi: 10.1016/S2213-2600(18)30184-X. Epub 2018 May 20. PMID: 29792288.
Revisor
Francisco Javier Callejas González.
Servicio de Neumología. Complejo Hospitalario Universitario de Albacete (CHUA).
Tema: Eficacia y seguridad del tralokinumab en el asma grave no controlada.
Palabras clave: Tralokinumab; IL-13; anticuerpo anti-IL-13; asma grave no controlada; exacerbaciones.
Resumen
El tralokinumab es un anticuerpo monoclonal humano anti-interleucina-13 (IL-13) desarrollado para el tratamiento del asma grave no controlada. El programa ATMOSPHERE de desarrollo clínico del tralokinumab consta de cinco ensayos clínicos (EC):
- Dos pivotales de fase 3, STRATOS 1 y 2, con el objetivo de evaluar su eficacia y seguridad en el asma grave no controlada.
- Un ensayo clínico en fase 2, MESOS, para valorar el efecto del anti-IL-13 sobre la inflamación eosinofílica en pacientes asmáticos.
- Un ensayo clínico de fase 3, TROPOS, sobre el ahorro de corticoides sistémicos.
- Un ensayo abierto a largo plazo en participantes japoneses.
Este resumen tiene como objetivo comentar los resultados obtenidos en los ensayos STRATOS 1 y STRATOS 2
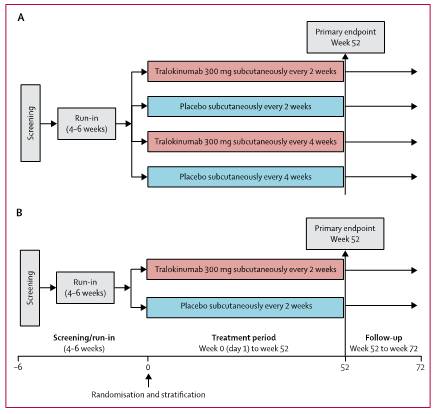
STRATOS 1 y 2 eran EC, doble ciego, de grupos paralelos, controlados con placebo, con participantes de 12 a 75 años de edad con asma grave no controlada a pesar del uso de corticosteroides inhalados (≥ 500 μg por fluticasona o equivalente) y un agonista β2 de acción prolongada. El STRATOS 1 se realizó en 246 centros de 14 países, y el STRATOS 2, en 242 centros de 13 países. En STRATOS 1, los participantes fueron asignados al azar (2:1) para recibir 300 mg de tralokinumab o placebo de forma subcutánea cada 2 semanas o cada 4 semanas durante 52 semanas, y en STRATOS 2 fueron asignados de manera aleatoria (1:1) para recibir tralokinumab 300 mg o placebo de forma subcutánea cada 2 semanas durante 52 semanas. El STRATOS 1 intentó identificar población con respuesta positiva de algún biomarcador en cuanto a la utilización del tralokinumab; y posteriormente el STRATOS 2 comprobó la respuesta del tralokinumab en la población identificada con biomarcadores positivos en STRATOS 1. En la figura 1 puede verse el diseño de ambos EC.
Los biomarcadores valorados en STRATOS 1 fueron el recuento de eosinófilos en sangre, la fracción de óxido nítrico exhalada (FENO), dipeptidil peptidasa-4 en (DPP-4), periostina e IgE en suero.
La variable principal del estudio fue la reducción anual de la tasa de exacerbaciones de asma (AAER) en la semana 52 en la población de STRATOS 1, mientras que en STRATOS 2 se valoró la AAER en la población que presentaba un biomarcador predictivo con una mejor respuesta al tralokinumab. En ambos ensayos, las exacerbaciones del asma fueron definidas como un empeoramiento del asma que llevó al uso de corticosteroides sistémicos durante 3 o más días, una visita a urgencias que requirió uso de esteroides sistémicos o un ingreso hospitalario por este motivo.
Figura 1. Diseño de STRATOS 1 (A) y STRATOS 2 (B).
Otras variables de estudio fueron el cambio porcentual del FEV1 prebroncodilatador desde el inicio hasta la semana 52, tiempo hasta la primera exacerbación y síntomas, control de la patología y calidad de vida, entre otros (ACQ-6, AQLQ, EQ-5). Además, se comprobó el perfil de seguridad del tralokinumab en ambos EC, siendo registrados todos los efectos adversos (EA), incluidos los graves y aquellos que llevaron a la interrupción del fármaco, definidos los EA como los ocurridos dentro de las 72 horas desde la administración del fármaco en investigación.
Ambos ensayos se iniciaron en 2014, el STRATOS 1 en junio y el STRATOS 2 en octubre, y ambos finalizaron en 2017, en febrero y septiembre respectivamente. Entre ambos estudios se incluyó a más de 2.000 pacientes.
Respecto a los resultados obtenidos, en toda la población de STRATOS 1 el tralokinumab cada 2 semanas no redujo significativamente la AAER en comparación con el placebo (7% de reducción [95%, IC 20,8-28,4]; rate ratio 0,93 [95%, IC 0,72-1,21]; p = 0,59), pero sí se identificó a una población mejor respondedora, que fue aquella en la que los niveles de FENO eran ≥ 37 ppb, donde tralokinumab cada 2 semanas (n = 97) redujo el AAER en un 44% (IC 95%, 6-66; rate ratio 0,56 [95%, IC 0,34-0,94]; p = 0,028) en comparación con placebo (n = 102). Sin embargo, esta mejoría mostrada en STRATOS 1 no se confirmó en la población de STRATOS 2, en que en los pacientes con FENO elevado que recibieron tralokinumab cada 2 semanas (n = 108), la AAER no mejoró significativamente (15,8% de reducción [95%, IC 33,7-47,0]; rate ratio 0,84 [95%, IC 0,53-1,34]; p = 0,47) en comparación con placebo (n = 121).
En el subgrupo de participantes con FENO elevado (≥ 37 ppb) se observó un aumento significativo del FEV1 prebroncodilatador en los tratados con tralokinumab cada 2 semanas en comparación con el placebo (12,8% [95%, IC 5,34-20,26]; p = 0,00079), lo que se tradujo en una mejora clínicamente significativa del FEV1 absoluto de aproximadamente 340 ml en la semana 8 del inicio del tralokinumab, y se mantuvo hasta la semana 52 (figura 2).
En cuanto al perfil de seguridad, en el STRATOS 1 278 (69,8%) de los 398 participantes con tralokinumab cada 2 semanas tuvieron eventos adversos, en comparación con 243 (60,8%) de 400 en el grupo de placebo combinado, mientras que en el STRATOS 2 los eventos adversos ocurrieron en una tasa similar entre los grupos de placebo (290 [68,7%] de 422) y tralokinumab (306 [72,0%] de 425). Los eventos adversos más frecuentes fueron asma, cefalea e infección del tracto respiratorio superior, casi todos de intensidad leve o moderada y la mayoría no se consideraron relacionados con el fármaco en investigación. Respecto a los EA más frecuentes que se consideraron relacionados con el tratamiento con tralokinumab cada 2 semanas, en el STRATOS 1 fueron el eritema en el lugar de la inyección (en 24 [6%] de 398 participantes), dolor en el lugar de la inyección (en 15 [3,8%]) y reacción en el lugar de inyección (en 16 [40%]); en STRATOS 2, reacción en el lugar de la inyección (en 23 [5,4%] de 425 participantes) y eritema en el lugar de la inyección (en 15 [3,5%]).
Figura 2. Medida basal del FEV1 prebroncodilatador en STRATOS 1.

Finalmente, se concluyó que el perfil de seguridad del tralokinumab es aceptable, y que este fármaco redujo la AAER en pacientes con FENO ≥ 37 ppb en el STRATOS 1, lo que no pudo confirmarse en el STRATOS 2.
Comentario
En resumen, el tralokinumab no cumplió con el objetivo primario de reducir la AAER, ni en el STRATOS 1 ni en el STRATOS 2. Aunque sí es cierto que se identificó una población con mejor respuesta en el STRATOS 1, aquellos con FENO ≥ 37 ppb, donde sí se redujo la AAER, este efecto no pudo confirmarse en el STRATOS 2 (había un beneficio, pero no fue clínicamente significativo).
Lo que en un inicio se consideró una de las principales fortalezas de los ensayos STRATOS, a saber, el diseño escalonado, pues permitió la identificación de un subgrupo biomarcador positivo en el STRATOS 1 (FENO elevado) que se investigó en el STRATOS 2, probablemente resultó una limitación, pues la prevalencia de participantes con FENO elevado en el segundo ensayo, a pesar de ser coincidente con el STRATOS 1, fue inferior a la prevista originalmente, lo que podría haber afectado a la potencia estadística del STRATOS 2. Además, a medida que los ensayos avanzaron al mismo tiempo, no hubo oportunidad de aumentar la población del STRATOS 2 para un subgrupo de FENO elevado una vez se identificó dicho FENO como el biomarcador predictivo.
Así, los hallazgos obtenidos en estos ensayos se suman a la evidencia previa disponible de que el bloqueo único de la IL-13 parece insuficiente para reducir las exacerbaciones del asma en personas con asma grave no controlada, aunque sí podría mejorar la función pulmonar.